El síndrome de Peitz-Jeghers-Touraine es un trastorno genético poco común causado por una mutación del gen STK 11 en el cromosoma 19. El tipo de transmisión es autosómico dominante.
Historia
En 1921, el caso se describió por primera vezuna enfermedad que ahora se conoce como síndrome de Peitz-Touraine-Jeghers. La descripción fue realizada por el médico holandés Johannes Peitz, quien estudió el síndrome utilizando el ejemplo de una familia holandesa. En 1949, su investigación científica fue complementada por el médico estadounidense Harold Yegers.
Sintomatología
El síndrome de Peitz-Jeghers se caracteriza porproliferación de pólipos de hamartoma en el tracto gastrointestinal (de ahí el otro nombre de esta enfermedad: hamartoma poliposis). Se encuentran con mayor frecuencia en el intestino delgado o grueso, y con menor frecuencia en el estómago y el recto. Al principio, los pólipos no se hacen sentir, pero a medida que crecen, aparecen estreñimiento, hinchazón, sangrado rectal y dolor abdominal opresivo. Aproximadamente la mitad de los pacientes experimentan complicaciones en forma de obstrucción intestinal. El sangrado latente es peligroso porque puede provocar anemia y, como resultado, debilidad general, mareos y disminución del rendimiento. Los pacientes no prestan atención de inmediato a los síntomas característicos de la enfermedad: diarrea frecuente y dolor abdominal, ya que esto puede ser un signo de muchas otras enfermedades gastrointestinales. Pero el síndrome de Peitz-Jeghers puede tener consecuencias extremadamente graves en ausencia de tratamiento, por lo que no debe dudar en visitar a un especialista.
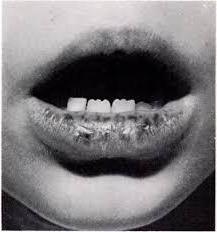
Las manchas pigmentadas en las membranas mucosas (labios, palmas, pies, membranas mucosas de los ojos) también indican la probable presencia del síndrome de Peutz-Egers.

Las manchas aparecen a una edad temprana, a veces enbebés lactantes. Son planas, de color marrón oscuro, con un tamaño de 1 a 5 mm. Con el tiempo, las manchas de los labios pueden aclararse y desaparecer, pero permanecen en la membrana mucosa de la boca para siempre. Este es un síntoma importante en el diagnóstico, ya que el 98% de los pacientes con síndrome de Peitz-Jeghers tienen pigmentación en las membranas mucosas.
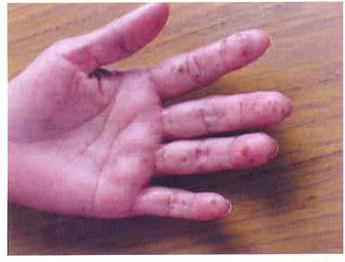
Dado que el síndrome de Peitz-Jeghers-Touraine esuna enfermedad genética, al hacer un diagnóstico, el especialista debe tener en cuenta el historial médico de todos los miembros de la familia. Si uno de ellos padecía esta enfermedad, entonces todos sus familiares tienen un gen mutado.
Diagnostico
Diagnostique el síndrome de Peitz-Jeghers usandobiopsia. Si se encuentra un componente de hamartoma en la parte del pólipo que se toma para el análisis, entonces este es un síntoma típico de esta enfermedad. Los pólipos que miden de 1 a 5 mm por lo general no interfieren con el funcionamiento normal del tracto gastrointestinal. Pero a medida que crecen, pueden provocar sangrado, porque los crecimientos de más de 1 cm deben eliminarse. Los pólipos se caracterizan por un crecimiento moderado, pueden ser múltiples y únicos. Con múltiples tratamientos, el tratamiento es mucho más difícil. No se pueden eliminar una vez, por lo tanto, se usa una dieta suave, así como una terapia con medicamentos destinados a retrasar el crecimiento de las neoplasias.
Otros criterios de diagnóstico importantes son:herencia y pigmentación mucosa. Dado que los pólipos se encuentran en pacientes mayores de 10 años, para los niños, la pigmentación en las membranas mucosas es el síntoma principal al momento de hacer un diagnóstico. El síndrome de Petz-Jeghers también suele ir acompañado del síndrome de McCune-Albright (desarrollo sexual temprano). Si un niño tiene un desarrollo pre-sexual, la probabilidad de desarrollar poliposis hamartoma es bastante alta.
Riesgo de cáncer
En pacientes con síndrome de Peitz-Jeghersel riesgo de desarrollar cáncer gastrointestinal es significativamente mayor que en personas sanas. Muy a menudo, la oncología afecta el intestino grueso, el intestino delgado y el páncreas. En las mujeres, a menudo se observa cáncer de mama (45% de los casos). Es en la alta probabilidad de desarrollar cáncer donde reside el peligro de la enfermedad; el síndrome de Peitz-Jeghers debe controlarse mediante varios procedimientos clínicos.
Dado el mayor riesgo de desarrollar oncología, todos los pacientes deben someterse a una radiografía del intestino delgado aproximadamente una vez cada 2 años.

Las mujeres mayores de 25 años deben realizarse una mamografía todos los años para detectar el cáncer en una etapa temprana.
En el diagnóstico de cáncer, un estudio de la presencia de antígeno carcinoembrionario en la sangre juega un papel importante. Es una sustancia química que se produce en el cáncer de intestino, de mama o de pulmón.
La determinación de otros antígenos del cáncer también es importante.(también son antígenos tumorales): sustancias producidas en las células afectadas por el cáncer de cualquier órgano. Su naturaleza depende de la localización del tumor, por lo que estas sustancias juegan un papel importante en el diagnóstico.
La mayoría de los pacientes con síndrome de Peitz-Egers novivir hasta 60 años, muriendo de cáncer de páncreas (11%), estómago (57%), intestinos (85%), mama (45%). El riesgo de desarrollar oncología de los pulmones, testículos, cuello uterino y ovarios también aumenta ligeramente. Si la oncología se detecta en una etapa tardía, es fatal. Por eso es tan importante el diagnóstico oportuno.

Tratamiento
A pesar de que ya en 1921 Peitz describióSíndrome de Peitz-Jeghers-Touraine, los síntomas y el tratamiento de esta enfermedad aún no se comprenden bien. Por el momento, no se ha desarrollado un tratamiento integral para esta enfermedad. Dado que con el síndrome crecen cientos de pólipos en los intestinos, es imposible eliminarlos todos de manera profiláctica debido al alto riesgo de tal operación. Por lo tanto, los pacientes con síndrome de Peitz-Jeghers durante su vida se someten a muchas intervenciones quirúrgicas para extirpar los pólipos cuando alcanzan un tamaño crítico.
El resultado
Aunque completamente curado de esta enfermedad eneste momento es imposible, la terapia con medicamentos ralentizará significativamente el crecimiento de los pólipos, normalizará los intestinos y el estómago. El diagnóstico temprano del cáncer también aumenta las posibilidades de cura después de la extirpación del tumor.








